
Portable Nanopore Sequencing Technology Enables Fast, Accurate and Cost-effective Genetic Testing
The emergence of high throughput Next Generation Sequencing (NGS) has transformed biomedical studies and catalyzed novel practices of genetic testing. Currently, NGS is the most commonly used sequencing platform due to its affordability and read accuracy. One of the biggest disadvantages associated with NGS is its short read length, ranging from 100-300 bps, preventing its effective use in de novo genome assembly and detection of genome structural variations. In addition, heavy reliance on instrumentation makes NGS not suitable for portable or onsite detection of DNA. Portable DNA sequencing technology with ultra-long read length is in desperate need for fast genetic testing, de novo genome assembly and genome finishing, and for the discovery of structural variants, especially for those in a relatively large genome such as the human genome.
Technology designed to generate long reads is referred to as third generation sequencing, an example of which comes from Oxford Nanopore Technologies. The Nanopore sequencing has recently been launched and has been used for pathogen detection or genome improvement. It can generate reads up to 1M bps in size with roughly 20 Giga bps in yield per run. Importantly, its ease of library preparation and portability make it a good choice for third generation sequencing. In addition, the technology offers direct sequencing of DNA and RNA without any labelling or amplification step. This holds great promise for the identification of base modifications in DNA or RNA. We have recently applied the Nanopore technology and generated genome assembly of C. nigoni with chromosomal contiguity. However, the technology is still under active development. For example, it’s read accuracy is approximately 85% to 92%. Its ultra-long reads are extremely useful for resolving the sequences in the “difficult” genomic regions that are highly repetitive, and for the detection of structural variants spanning large genomic intervals. This potential depends on a reliable and robust computational pipeline that enables fast and accurate identification of genomic variants. Unfortunately, existing pipelines for identification of genome variants using the long reads are not only computationally expensive, but also are cost ineffective. A fast, accurate and cost-effective pipeline is in need for taking full advantage of the Nanopore long reads, especially for rapid genetic testing. We therefore propose to develop an application platform by taking advantage of Nanopore sequencing, which has implications not only in precision medicine, but also in cost-effective and real-time genetic testing, including detection of transgenes in genetically modified organisms.
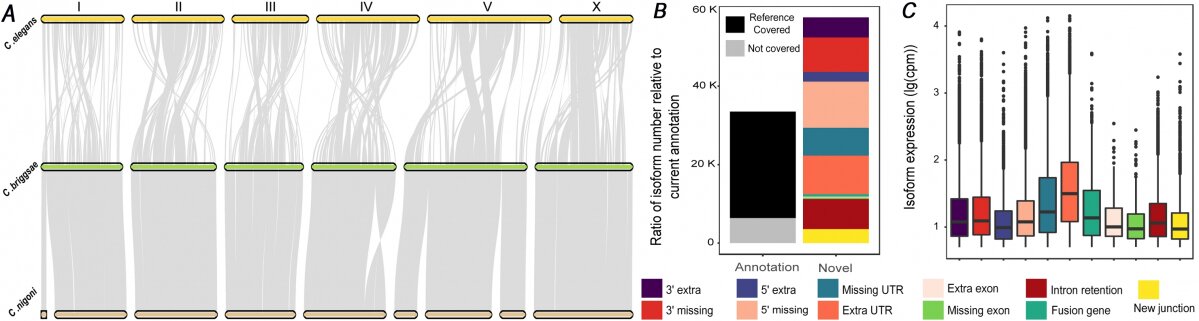
Figure 1. Use of sequences of paired ends of genomic constructs and Nanopore DNA sequencing in genome assembly (A) or Nanopore Direct RNA sequencing in identification of novel transcript isoform (B & C).
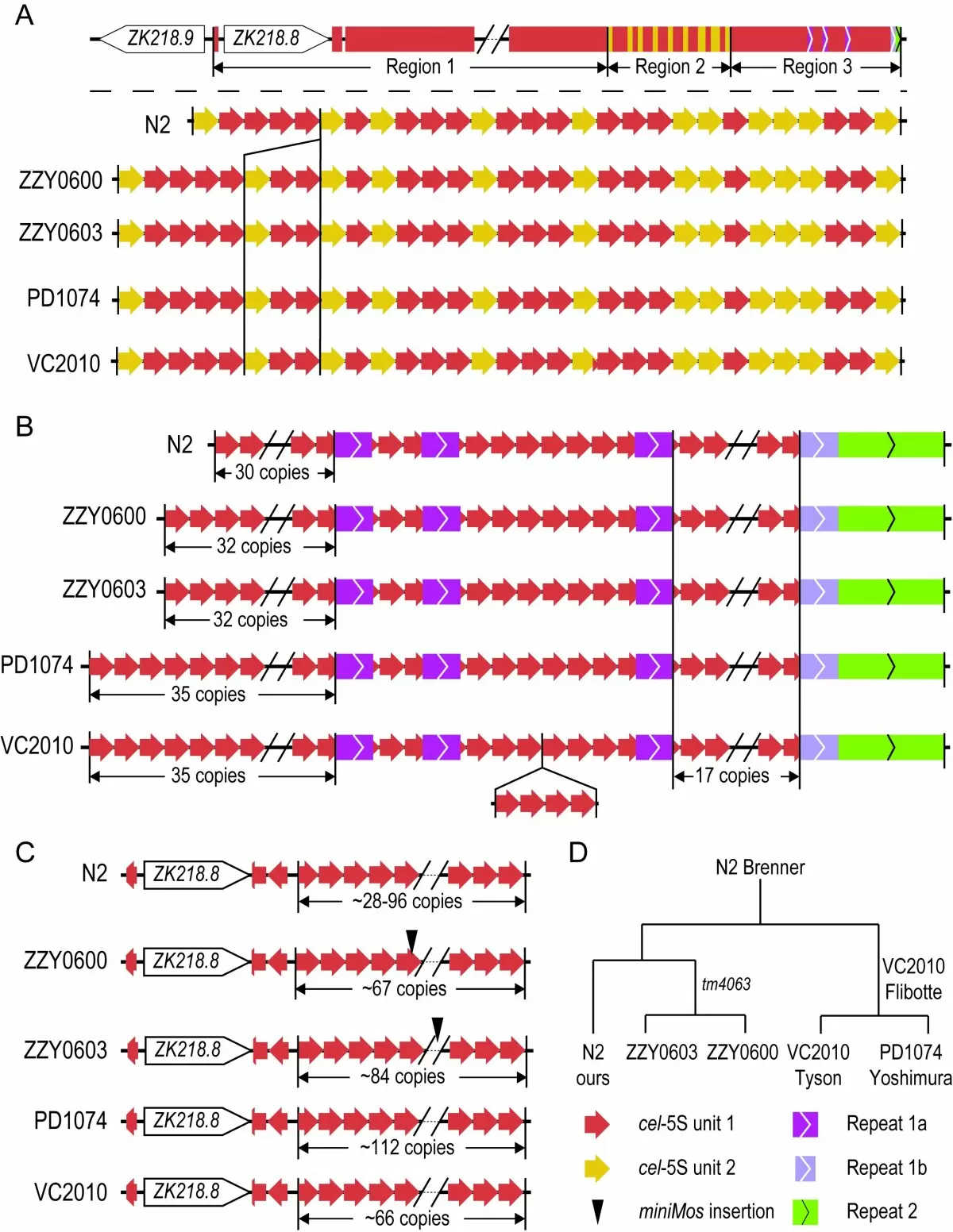
Figure 2. Unit number dynamics within the 5S rDNA cluster among our N2 and other N2-derived C. elegans strains using Nanopore DNA sequencing.
(A) Comparison of unit compositions in the Region 2 of 5S rDNA cluster between strains. Overview of 5S rDNA cluster is shown on the top and comparison of the Region 2 between strains on the bottom. Note that there seems a deletion of 3 units (highlighted), including two copies of unit 1 and one copy of unit 2, in our N2 when compared with the remaining strains (See (D) for strain origin).
(B) Comparison of unit compositions in the Region 3 of 5S rDNA cluster between strains. Unit copy number shows the highest conservation on the right arm (17 copies in all strains). An insertion of 4-copies of unit 1 in VC2010 is highlighted. The left-arm shows the highest dynamics in the rDNA copy numbers ranging from 30 to 35 among strains.
(C) Comparison of unit compositions in the Region 1 of 5S rDNA cluster consisting of unit 1 only between strains. Dynamics of the uncertain copy numbers of unit 1 are due to a lack of reads spanning the entire region. Insertions of our transgenes are indicated with a triangle.
(D) Origins of the strains used above. Our N2 is shipped from Waterston lab, Seattle, WA, USA in 2010. PD1074 is a recent derivative of VC2010. ZZY0600 and ZZY0603 were generated by transgene insertion into unc-119(tm4063) worms, which was derived from C. elegans N2.
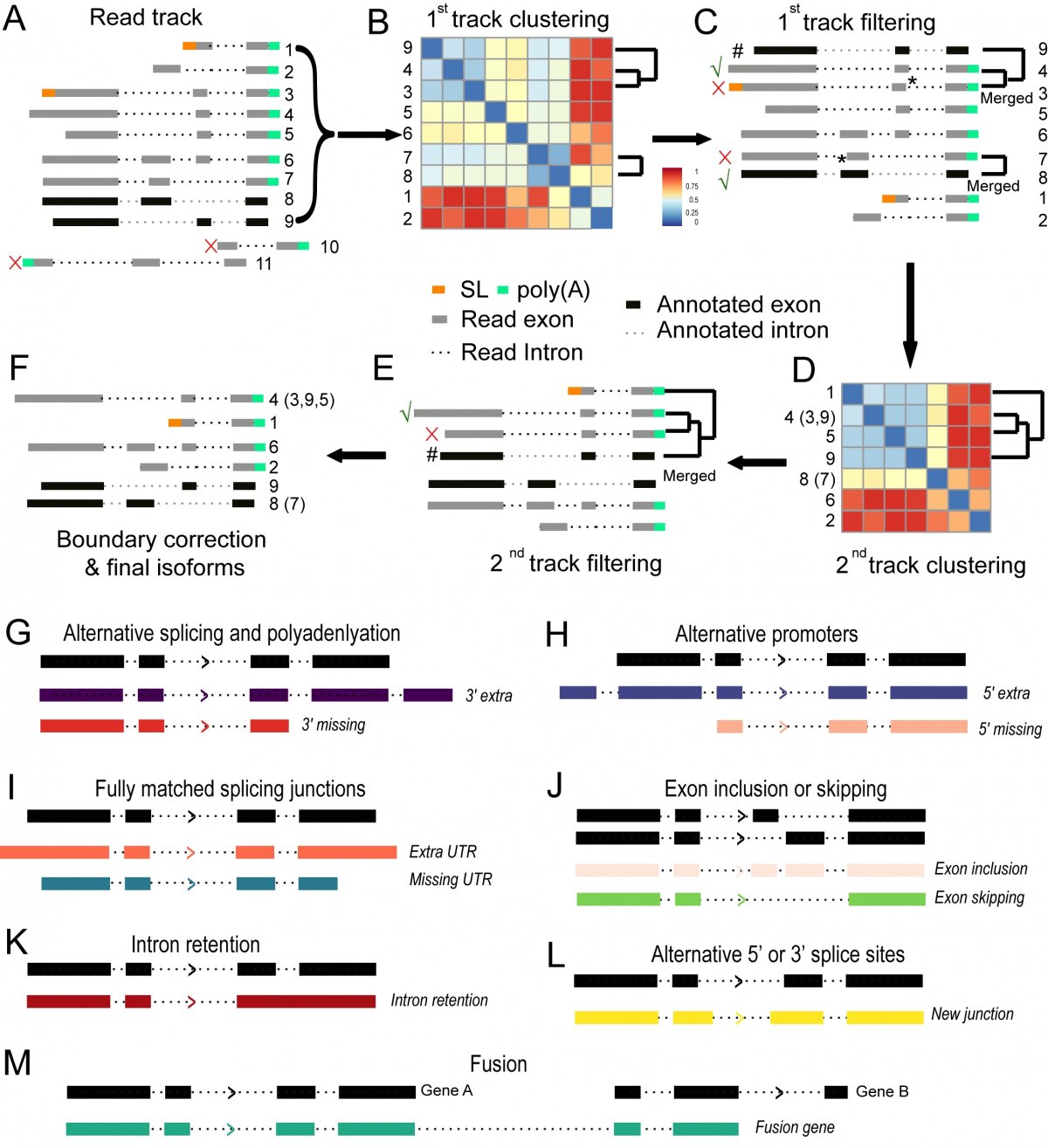
Figure 3. Flowchart of new isoform calling from Nanopore RNA sequencing reads by TrackCluster.
(A) Assignment of sequence tracks (shown as grey bar with different numbers) to a given locus based on read mapping against the C. elegans genome. Existing isoforms are included as individual track (black bar). Two reads that show few overlap with any existing exon or are antisense are excluded from subsequent analysis for the current locus.
(B) The 1st round of clustering of tracks based on their distance scores (see Materials and Methods).
(C) Read tracks (excluding existing transcript) are merged if their distance scores satisfy our cutoff. Only the one with the biggest size of summed exons is retained (indicated with “”) from each group along with the existing one (indicated by #) for the subsequent isoform calling. The remaining tracks (indicated with “x”) including existing transcript are assigned as “subread” and used only for expression quantification and boundary correction. Note, during track merging, a minor shift (indicated with “*”, within 5% change in “score 1” defined in Materials and Methods) in exon-intron boundary caused by read error is permitted to avoid over-calling of novel isoform.
(D) The retained tracks from (C) are subjected to the 2nd round of track clustering based on mutual distance scores (see “score 2” in Materials and Methods).
(E) The tracks (including existing transcript) are merged if their distances satisfy our cutoff to avoid calling a novel isoform for a possible partially degraded read retained in (C) except those starting with an SL.
(F) Existing annotated (black) and novel isoforms (grey) after junction correction (see Fig. S2). The retained track is called a novel isoform due to its distance score with any existing transcript satisfying our cut off.
(G-I) Schematic representation of each category of the newly identified isoform. Novel isoforms involving newly defined 5’ or 3’ end. “5’ or 3’ extra or missing” is defined as novel isoform with extra or missing exon at either end of a novel isoform relative to an existing transcript. “Extra or missing UTR” is defined as novel isoform involving changes only in UTR relative to an existing transcript.
(J-M) Novel isoform involving exon change within gene body.
Research Output
- A. Runsheng Li, Xiaoliang Ren, Qiutao Ding, Yu Bi, Dongying Xie, Zhongying Zhao*. Direct full-length RNA sequencing reveals unexpected transcriptome complexity during C. elegans development. Genome Research. 2020 Feb;30(2):287-298.
- Shoudong Zhang, Runsheng Li, Li Zhang, Shengjie Chen, Min Xie, Liu Yang, Yiji Xia, Christine H. Foyer, Zhongying Zhao*, Hon-Ming Lam* New insights of Arabidopsis transcriptome complexity revealed by direct sequencing of native RNAs. Nucleic Acids Research. 2020,
https://doi.org/10.1093/nar/gkaa588. - Xiaoliang Ren, Runsheng Li, Xiaolin Wei, Yu Bi, Vincy Wing Sze Ho, Qiutao Ding, Zhichao Xu, Zhihong Zhang, Chia-Ling Hsieh, Amanda Young, Jianyang Zeng, Xiao Liu, Zhongying Zhao*. Genomic bases of recombination suppression in the hybrid between hermaphroditic Caenorhabditis briggsae and gonochoristic C. nigoni. Nucleic Acids Research. 2018 Feb 16;46(3):1295-1307.
- Runsheng Li, Xiaoliang Ren, Yu Bi, Vincy Wing Sze Ho, Chia-Ling Hsieh, Amanda Young, Zhihong Zhang, Tingting Lin, Yanmei Zhao, Long Miao, Peter Sarkies and Zhongying Zhao*. Specific Downregulation of Spermatogenesis Genes Targeted by 22G RNAs in Hybrid Sterile Males Associated with an X-Chromosome Introgression. Genome Research. 2016, 26(9):1219-32
* Corresponding author
Principal Investigator

Co-investigators

Co-investigators (Non-Lab Members)




Dr. Zhihong Zhang






