
Novel Algorithms for Constructing a High-resolution Global Human Gut Microbiota Catalog
Human gut microbiota has recently been recognized as our "second genome" that closely correlates with human health and drugs' effects. Knowing the genomic sequences of the microbiota content allows us to study its functions. However, microbial genome sequences are difficult to obtain. While a few microbes can survive after isolation and be cultured in vitro for sequencing, the remaining microbial content remains as "microbial dark matter". Current studies sequence a mixture of microbial genomes from human stool samples using short-reads, which are aligned to the "reference gene catalog" to quantify the gene abundances. But the results could be misleading because the same gene may belong to more than one microbe. We are eager to construct a high-resolution human gut microbiota catalog to transform the research from gene-centered to genome-centered. The limitation of short-read sequencing challenges the completeness and continuity of genome assembly, and the introduction of population sampling bias leads to an incomplete collection of reference genomes. For example, most of the published metagenome data is taken from Europeans and Americans, thus they are unable to reflect the characteristics of Asians due to the discrepancy of diet and lifestyles.
This project aims to construct a high-resolution global human gut microbiota catalog by collecting the individuals from diverse continents and countries. To achieve this goal, we breakdown the project into four subtasks:
1. Integrate available human gut microbial genomes.
We will develop a comprehensive integration pipeline to automatically download and collect the published microbial reference genomes from cultured and uncultured approaches.
2. Integrate and analyze the public and private raw microbiome sequencing data.
We will implement a computational framework to analyze microbiome sequencing data from two sources: 1) 11,400 participants collected from recent publications; 2) We also sequenced 4,830 metagenomes from Han Chinese stool samples.
3. Construct microbial phylogenetic trees and annotate novel genes.
The novel microbial species and genes will substantially expand our understanding of the human gut microenvironment.
4. Develop novel metagenome assembly algorithms for long-fragment sequencing.
As a supplement to short-read sequencing, we will develop novel algorithms to reconstruct high-quality microbial genomes using long-fragment sequencing.
We believe this project will bring a significant breakthrough to the field by contributing new data, new resources and new algorithms. Our human gut metagenome catalog would benefit all subsequent analysis and shed light on microbiota-targeted disease therapies and drug discovery.
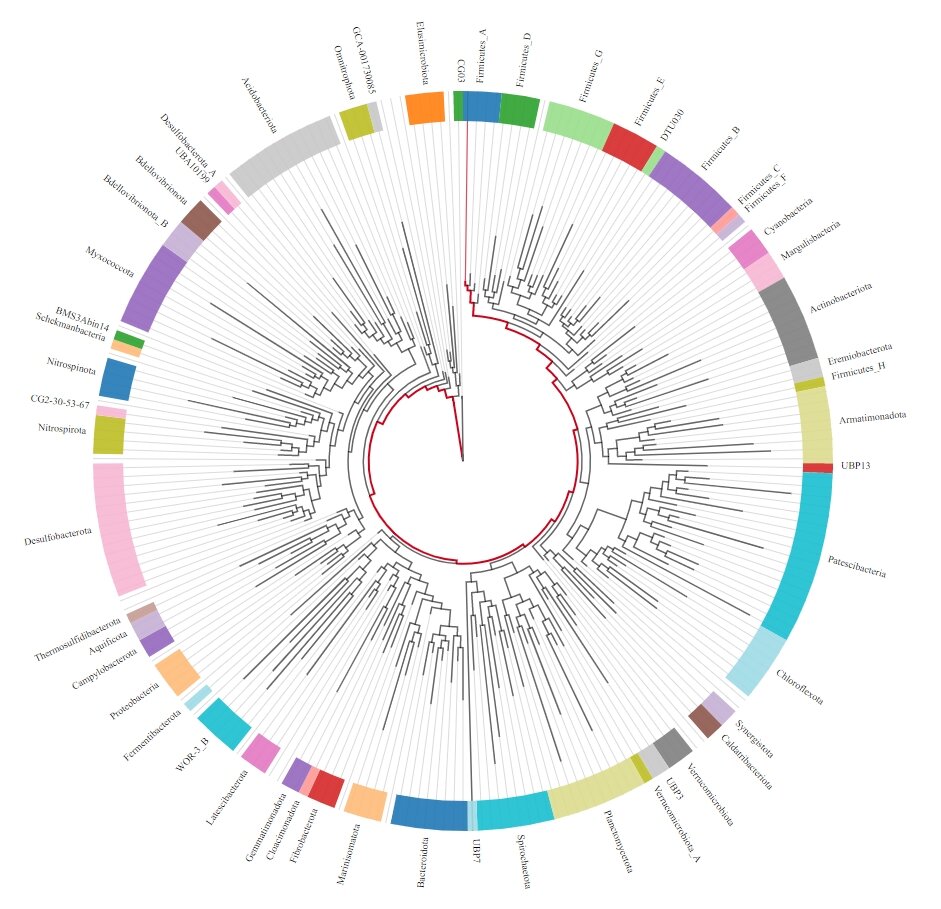
Figure 1.
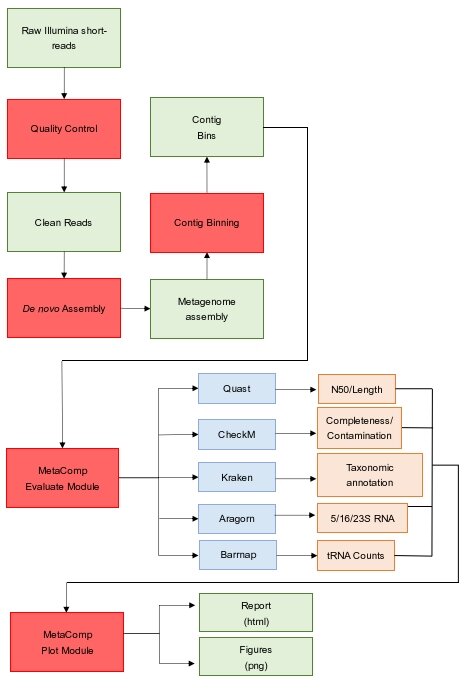
Figure 2.
Related Publications:
- Lu Zhang, Zhenmiao Zhang, Xin Feng, Yang Chen, Xiaodong Fang. A high-resolution global metagenome catalog from uncultured human gut microbiota, in preparation.
- Lu Zhang, Xiaodong Fang, Herui Liao, Zhenmiao Zhang, Yen Kaow Ng, Xin Zhou, Lijuan Han, Yang Chen, Qinwei Qiu, Shuai Cheng Li. A comprehensive investigation of metagenome assembly by 10x linked-read sequencing, under review.
- Zhenmiao Zhang, Jing Zhang, Xiaodong Fang, Lu Zhang. A comprehensive evaluation for multi-platform metagenome assembly, in preparation.
- Xin Zhou, Lu Zhang, Ziming Weng, David L. Dill, Arend Sidow. Aquila: diploid personal genome assembly and comprehensive variant detection based on linked reads, under review. https://www.biorxiv.org/content/10.1101/660605v2
- Xin Zhou, Lu Zhang, Xiaodong Fang, Yichen Liu, David L. Dill, Arend Sidow. Aquila stLFR: assembly based variant calling package for stLFR and hybrid assembly for linked-reads. https://www.biorxiv.org/content/10.1101/742239v2
- Lu Zhang, Xin Zhou, Ziming Weng, Arend Sidow. De novo diploid genome assembly for genome-wide structural variant detection. NAR Genomics and Bioinformatics 2020 doi: 10.1093/nargab/lqz018.
- Lu Zhang, Xin Zhou, Ziming Weng, Arend Sidow. Assessment of human diploid genome assembly with 10x Linked-Reads data. GigaScience 2019 doi: 10.1093/gigascience/giz141.
Principal Investigator

Co-investigators

Co-Investigators (Non-Lab Members)

Prof. Xiaodong FANG (External)




