
Detecting the Tipping Points from Inflammation to Tumorigenesis using a Multi-level Network Modeling Approach
This project aims to detect the tipping points of cancers transiting from inflammation to tumorigenesis using genomic data. We will develop novel machine learning methods to reconstruct and characterize the structural changes in the GRNs that are closely related to the irreversible tipping point in inflammation-tumor translation, where a number of computational challenges are to be addressed. In particular, (i) effective reconstruction of GRNs based on the gene expression should be able to avoid capturing indirect influence among the genes to be the direct ones, which is non-trivial. (ii) GRNs are cell-type specific rather than uniform across all cells. Applying the existing methods for bulk RNA-seq can only capture the aggregated patterns of some dominating cell types, resulting in imprecise gene interaction characterization. (iii) Although there exist single cell RNA-seq (scRNA-seq) which can capture the gene expression in each cell, there are still technical limitations where many transcripts cannot be well-covered and thus their gene expressions become unexpected zeros. These dropout events make the existing GRN algorithms inapplicable to scRNA-seq data. (iv) The GRNs evolving over different disease stages, and thus their reconstruction, should be considered in a coordinated manner for better modelling the tipping points.
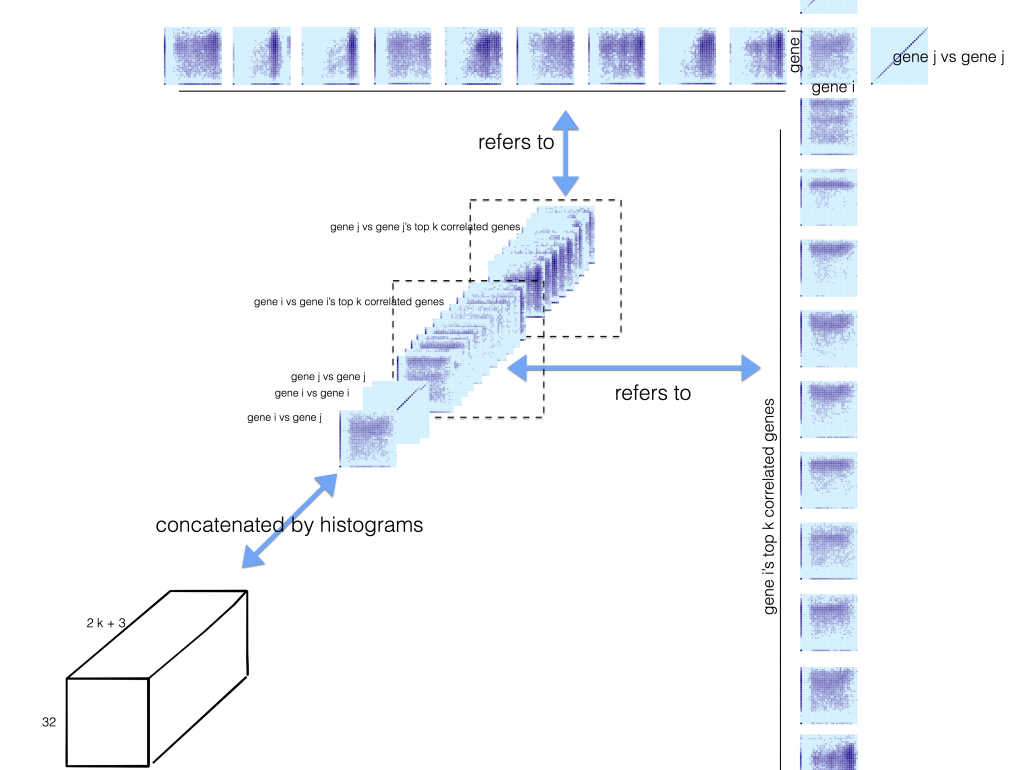
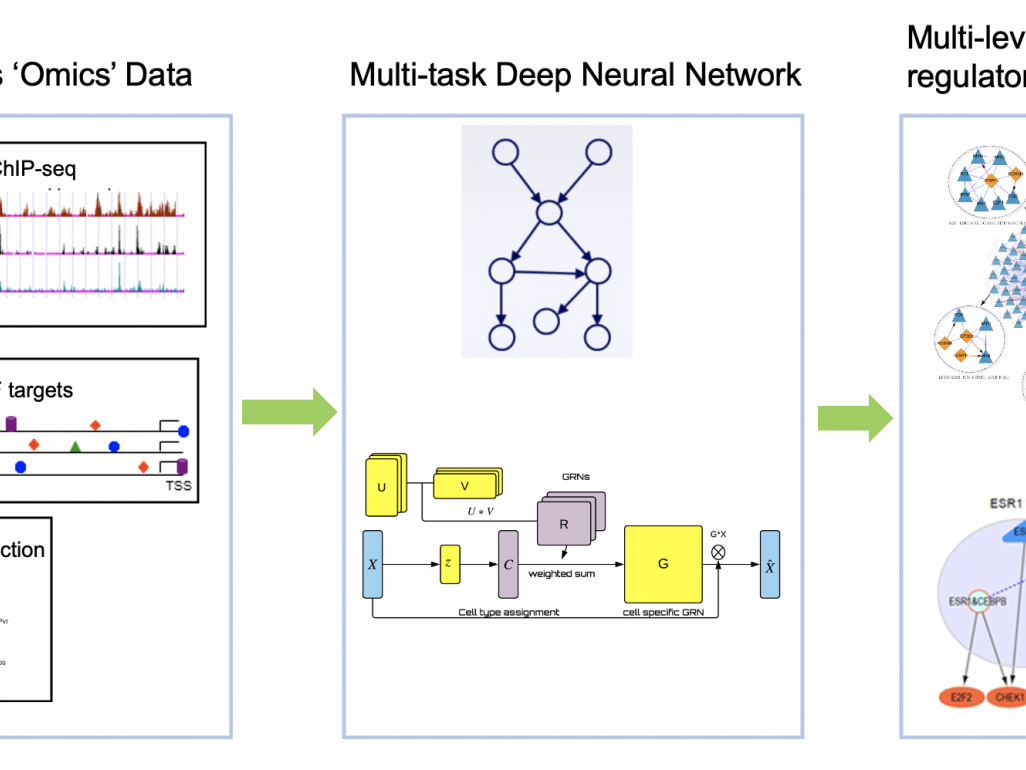
To address these challenges, we first propose a HSIC lasso-based regression method for GRN reconstruction, which can effectively avoid the indirect gene interactions. Also, we propose a convolutional neural network which is specific for the reconstruction from scRNA-seq by representing gene pair expressions as an image, and removing transitive interactions by involving multiple neighbouring gene pairs. This method requires a subset of regulatory relationships (ChIP-seq) to be available for supervised learning. To show their effectiveness, we applied them to public scRNA-seq data (including hepatitis and liver cancer) and found they significantly outperformed the state-of-the-art methods. To deal with situations lacking the labels for supervision, we propose an unsupervised multi-task deep neural network to integrate batch effect removal, dropout imputation, cell-type clustering and GRN reconstruction in a unified model to address all four challenges. The proposed models will also be extended to infer the evolving GRNs and/or cell type composition at different disease stages by exploring their commonality and specificity to pinpoint the key transcription factors for triggering disease transition. We believe our efforts could shed light on the molecular basis of gene therapy and drug discovery, and open a door to apply machine learning models to human genomics.


Research Output
- Jiaxing Chen, Chin Wang Cheong, Liang Lan, Aiping Lyu, William K. Cheung*, Lu Zhang*, Reconstructing cell type specific gene regulatory networks by convolutional neural networks, in preparation.
- Chin Wang Cheong, Jiaxing Chen, Liang Lan, Aiping Lyu, William K. Cheung*, Lu Zhang*, An integrative multi-task deep neural networks for single cell data analysis, in preparation.
- Lu Zhang, Jiaxing Chen, Shuai Cheng Li. A multimodal framework for detecting direct and indirect gene-gene interactions from large expression compendium. bioRxiv 2019 doi: https://doi.org/10.1101/680116.
- Lu Zhang, Cheng Qin, Junpu Mei, et al. Identification of microRNA Targets of Capsicum spp. using MiRTrans – a trans-omics approach. Frontiers in Plant Science 2017 doi: 10.3389/fpls.2017.00495.
- Yao Gao, Kexin Wang, Peng Wang, Junsheng Tian, Jingjing Chen, Boya Zhou, Daogang Guan, Xuemei Qin and Aiping Lu, A novel network pharmacology strategy to optimize components and decode mechanism of Lang Chuang Wan in treating systemic lupus erythematosus, submitted to Frontiers in Pharmacology.
- Kexin Wang, Yao Gao, Cheng Lu, Yao Li, Bo-Ya Zhou, Xuemei Qin, Guanhua Du, Li Gao, Gang Dao Guan and Aiping Lu, Uncovering the Complexity Mechanism of Different Formulas Treatment for Rheumatoid Arthritis Based on a Novel Network Pharmacology Model, submitted to Frontiers in Pharmacology.
Principal Investigator





Co-Investigators (Non-Lab Members)

Dr. Daogang GUANG (External)




